
The healthcare industry also called the medical industry is the scope of organizations and non-benefit organizations that give medicinal services, medical device manufacturer etc. The healthcare industry is going through a transformation, and to achieve success in this growing competitive environment, organizations need to make notable investments in processes and technologies to improve medical care. It involves the generation and introduction of products and services lending themselves to keeping up and restoring health.
The advanced medicinal industry is separated into numerous areas and relies upon interdisciplinary teams of trained professionals to address medical problems of people and populations. The medicinal industry is one of the world\’s biggest and quickest developing industries. Utilizing more than 10 percent of (GDP) of most created countries, health care can appear in a huge portion of the nation\’s economy.
- Operon Strategist is FDA 510 k process consultant helps the clients to register SBU (Small Business Unit), if applicable. Take out the testing requirement of the product, creation of the dossier, resolving the queries and after completion of all the activities, the client receives the US FDA 510 k approval.
- We also assist with the establishment registration and device listings to make suitable the supply of medical devices in the US.
In the medical industry, designing a medical device is the critical part for every manufacturer from the procedure of making the device as per the standard law until the trading of their device. Every medical device includes devices like implants, instruments, equipment to be used for treatment, monitoring and diagnostics. These devices also involve reconstructive devices such as hip and knee replacements and also implantable monitors for cardiac and diabetic care.
In the medical field, numerous technical terms, are included for the medical device in the form of documents etc, which every medical device manufacturer should know. In the medical device process, it consists of three types of records particularly that creates confusion to the new manufacturer who enters the medical device field: the design history file, 510(k) submission, and the technical file.
FDA 510 k Clearance & Premarket Approval for Medical Device
Operon Strategist is FDA 510 k process consultant helps the clients to register SBU (Small Business Unit), if applicable. Take out the testing requirement of the product, creation of the dossier, resolving the queries and after completion of all the activities.
Contact Us WhatsApp
https://www.youtube.com/watch?v=GVhCYVGhk60Similarly any sort of documents in medical device development, it requires a lot of effort. But in spite of that if you have good knowledge of medical device development, then it will be earlier for you and for every device manufacturer to find that there are similarities between each.
Design History File vs 510(k) vs Technical File
The design history file is documentation that mentions the process of a new device with its finished device design history. The design history file is an FDA term described in 21 CFR Part 820.30, which discusses design controls and how they should be kept in a design history file (DHF). This is basically the gathering of records from the design and development process.
As per the FDA, \”Every manufacturer should build-up and keep up a DHF for each sort of device. The DHF will include or reference the records important to exhibit that the design was developed with the approved design plans and the necessities of this part.\”
Some years back, DHF was in fact just a necessity characterized by FDA. ISO 13485:2003 made no direct said of a DHF, or something comparative. Even so, the latest updated ISO 13485:2016 currently indicates the need to build up \”design and development files \”.
The DHF presently has more importance since it is recognized as a key record containing design control documentation. This is the major part as because your design controls are your records exhibiting your device/product is safe.
Keeping up a DHF is most generally an extremely manual process, including old three-ring folder, file organizers, and paper. With the electronic-based quality management system, the making of the DHF has become much a much easier task.
As discussed above the DHF which has made you think that DHF is not a critical process for every device maker, Now the further topic is about the 510(k) and technical files. As DHF and your design controls feed into 510(k) & technical files.
Benefits of DHF
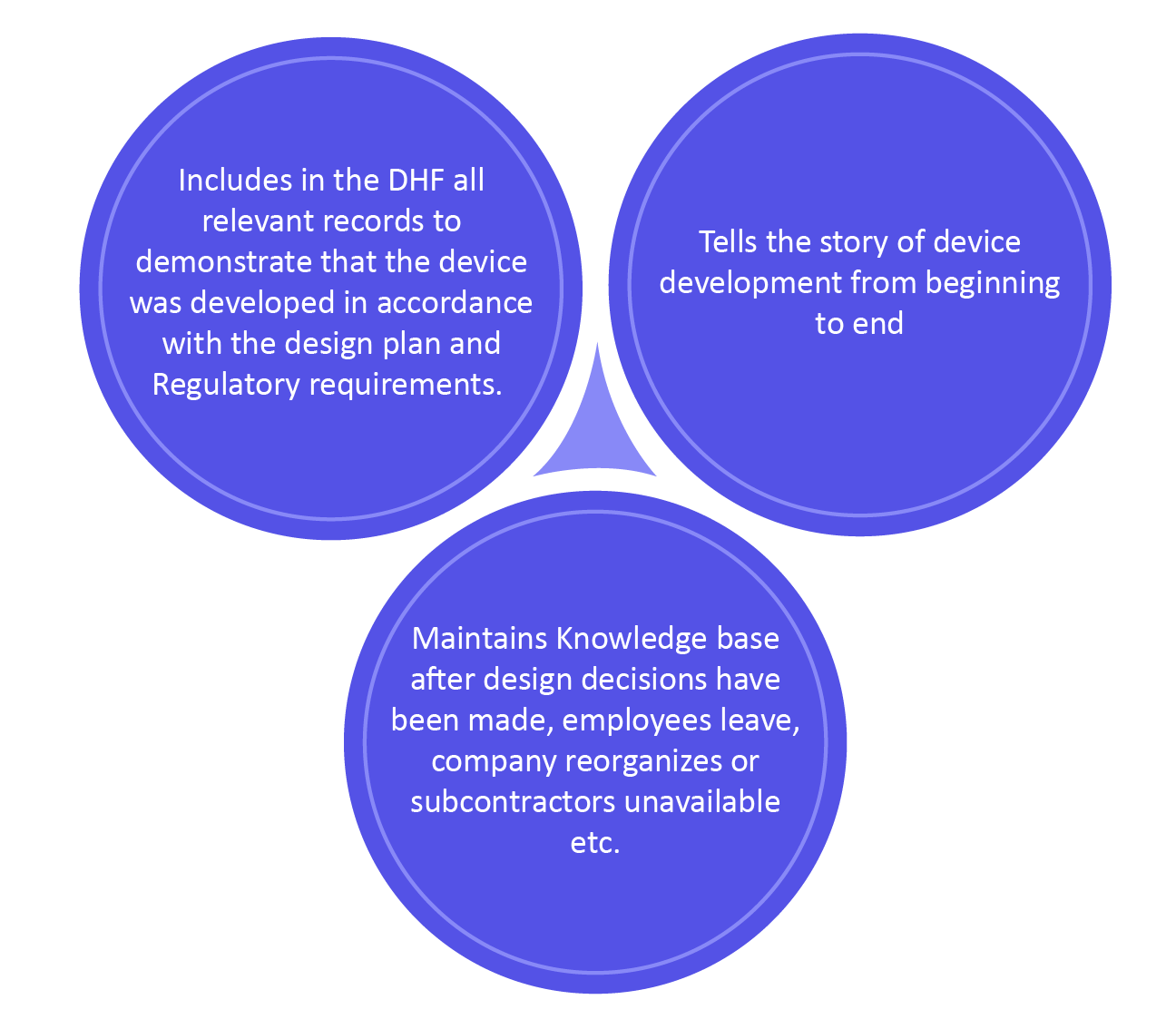
The 510(k) is not a form but actually it is a section of Federal Food, Drug, and Cosmetic Act (FD&CA, the Act), it is used for trading your device in simple words it is actually to obtain marketing clearance for a device. The 510(k) submission is the normal way for medical devices to pick up showcase leeway from FDA. A 510(k) is required for most Class II gadgets (and some Class I and Class III; some Class II don\’t require 510(k)). The goal is to exhibit to FDA that your device is generously identical to a predicate device, a product that has been already cleared by FDA. The important part of the 510(k) is to likewise exhibit your device is protected and works fine. Your design controls will sustain into a significant number of this area. It consists of device description, indications of use and performance so totally it contains 20 segments.
The 510(k) Submission
There are a lot many companies which couldn’t provide good reports of their company’s established design controls and also design history file. This ought to be your initial step before starting with your 510(k) as this process provides documentation report in the form of evidence of your finished product/device which actually proves that your device is safe for the patients and is effective and powerful too.
During the 510(k)process, if the FDA receives the details of the product which was described in the PMN (Premarket Notification) that says the device is similar to a medical device which is already available in the market. This allows the device manufacturer to sell their product without providing more information or testing (With the help of the device information which is similar in the market, the manufacturer receives 510k clearance). Once the 510(k) clearance is received, the device manufacturer can trade its device into the market, without passing the premarket approval (PMA) process. Your design controls and DHF will be an essential subject to their examination and you can finish up with 483 observations, and conceivably a notice letter if these aren’t decent.
The Technical File
A technical file is considerably likewise to a 510(k) than a design history file. This technical file is a collection of information in the form of documents that consists of details about the product and also that the product was designed as per the requirements of the Quality Management System (QMS). Every finished product/device that has a CE mark, it is compulsory for that product to have a technical file which consists of data that gives evidence about the product conforms with the EU directives for CE marked products. It is necessary to get your device into Europe and a few different parts of the world. The purpose of the technical file is to provide the conformity of the product to the applicable requirements and EU medical device (or IVD) regulations.
The technical file is somewhat less prescriptive than a 510(k) yet has a normal configuration. To assemble it, you ought to be through design verification and design validation first, whereas you just need to be through verification for the 510(k). Design validation might be addressed by comparative analysis, simulated use, animal studies, and/or clinical investigations.
A technical file is usually specified on the document archive system that occurs for a long existence, which is either done on a paper or in electronic files. This technical file includes device designs/drawing, specifications, reports, review records, instructions etc. One major difference with a technical file versus a 510(k) submission is the need to give a clinical evaluation report. A clinical evaluation report tends to the products clinical investigation, risk, and designs any post-market clinical follow-up (PMCF) studies that might be required. It is equivalent to and serves a comparative need to design validation.
Keep in mind:
- Determine your device classification early
- Establish good design controls
- Digitize your design history file for easy updating
- Establish your device master record
Our service range includes CE Mark certification consultancy services also our services include design history plan and technical file which we provide as per the companies requirement. A product must have a CE mark which declares that the product meets the EC directives. We are leading medical device CE Marking and other Regulatory Certification Consultant service provider for Medical Device Manufacturers dealing with disposable implant & syringe, surgical Instrument, orthopaedic implants etc. If required any assistance and more information contact us.


