
These controls should provide for an iterative design process with regular checks against specifications and relevant regulations, meaning that you can reduce the risks of omission and the amplification of mistakes as you go along. In fact, this iterative phased approach is not only the best commercial practice, it is a legal requirement. The FDA, the MHRA, ISO 13485 and all require that Design controls for medical devices is undertaken like this meaning that you cannot legally launch a device in any major market without proving that you have been working in this way.
Design Control
The term design controls originate from FDA regulations but it is as well mentioned in the ISO 13485.
There almost the same terms are used.
But whether you follow the FDA or the ISO requirements both ask for a complete documentation throughout the whole process and very similar to each other. The main goal is to establish and maintain procedures to control design of the device.
FDA Design Controls for Medical Devices
The Design control for medical devices follows a set of practices and procedures that help medical product developers:
- Manage quality.
- Ensure each product meets all requirements.
- Prevent potential issues or recalls in the future.
Medical design control stages from both the FDA and the ISO consist of:
- Design & development planning.
- Design inputs.
- Design outputs.
- Design review.
- Design verification.
- Design validation.
- Design transfer.
- Design changes.
- Design history file.
The regulations define each stage in a linear fashion. But each requirement is actually a part of a dynamic process that can change and repeat. This is known as the design and development planning model.
Medical Device Design Control Process:
An introductory stage from which Design Control begins is Design Input advancement and endorsement, which comprises of device plan and assembling procedures to be completed in the generation stage.
Configuration control is an all-encompassing methodology and doesn\’t end with exchanging the plan to the creation stage, once the outline is settled. It additionally impacts fabricating forms as indicated by the adjustments in the outline stage or even after generation input. It is a continuous procedure to build up an item that is usable for a client and hence for the upgraded item, it considers progressive changes from use design and additionally dissecting fizzled items. The picture underneath portrays the medical device development process in the waterfall design process (design control process flow chart).
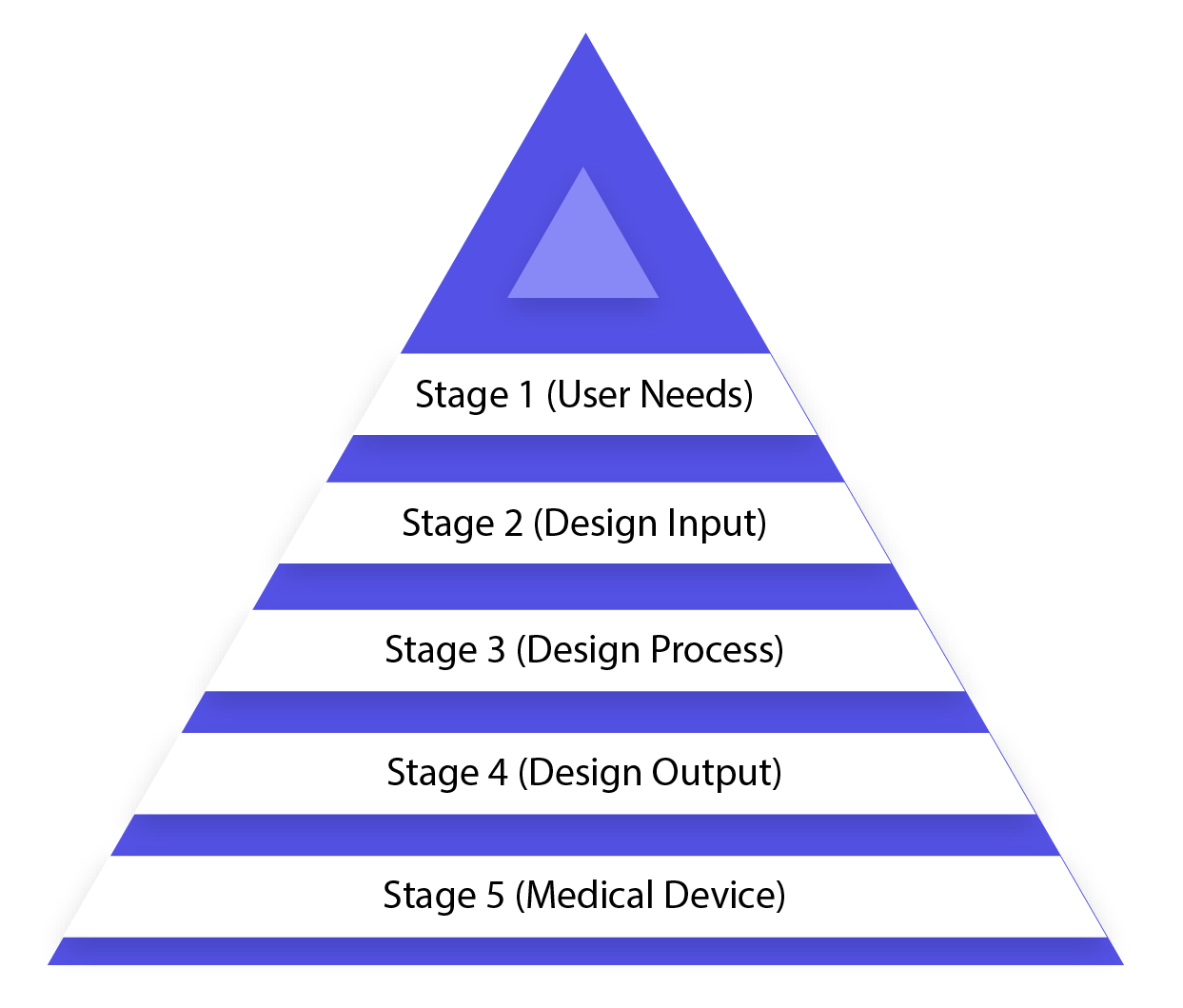
Stage 1 (User Needs)
Prerequisites are characterized considering the market requirements and the device is intended to address that need. After the arrangement of development, the medicinal device configuration is concluded and exchanged for creation for assembling. There is a need for input amid every single step of this procedure.
Stage 2 (Design Input)
This is an iterative procedure. At the point when an association chooses to address the specific need, the survey and test the adequacy of configuration input got from the need. By then, the iterative procedure of changing over prerequisites into device configuration begins.
Stage 3 (Design Process)
These outline inputs are changed over into configuration yield by changing over those prerequisites into abnormal state determinations (which are Design Output).
Stage 4 (Design Output)
Check process affirms whether the particulars are fulfilling prerequisites or not. Furthermore, the yield turns into the contribution to amend the necessities and this procedure goes ahead until the point that Design Output is lined up with the Design Input.
Stage 5 (Medical Device)
Once the last plan is prepared, it is transmitted to the generation office for mass assembling. Configuration control direction commands Design History File (DHF), which represents the linkages and connections between all the Design Control and help to follow all progressions all through the whole item improvement process. You can adopt a paper-based strategy or a product-based approach, particularly created for Design Control; your plan history document must be traceable and in addition available to all the colleagues.


